小分子药物设计、多肽生成、片段连接与逆折叠等任务,长期以来分别由不同模型与流程处理。小分子通常依赖图模型与3D生成框架,多肽设计则分为backbone构建、序列设计与侧链打包等多个阶段。不同任务拥有不同表示方式与训练数据,模型之间难以共享知识,也难以实现跨模态迁移。尽管近年来扩散模型和图生成模型快速发展,但“是否可以用一个统一模型同时处理多种3D分子生成任务”这一问题,一直缺乏清晰答案。
2026年2月21日,北京大学人工智能研究院博士生彭鑫港、助理教授张牧涵及合作团队在 Cell 期刊上发表论文Unified modeling of 3D molecular generation via atomic interactions with PocketXMol,针对“多模态分子生成长期分裂”这一核心问题,提出了一个基于原子相互作用的统一生成框架 PocketXMol。研究团队将小分子、多肽、片段链接、逆折叠等任务统一抽象为“在给定部分原子与键约束条件下补全剩余原子结构”的问题,在同一原子级表示空间内建模不同任务。模型无需区分分子类型,仅通过输入掩码控制生成区域,从而在结构引导小分子设计、3D分子生成、fragment linking、分子优化以及多肽设计等多种任务上取得稳定表现。
团队在 caspase-9 抑制剂设计与 PD-L1 结合肽设计中的实验进一步证明了模型生成分子具备真实生物活性。该研究提出了一种以“原子级统一表示”为核心的多模态分子生成范式,将原本彼此独立的设计问题整合为同一种条件生成框架,为跨分子类型的AI药物设计提供了新的方法基础。

图1. 论文截图
过去几年,AI 在分子设计领域的发展可以说是“多点开花”。
小分子生成有图扩散模型与3D构象生成框架;多肽设计有 backbone 生成、序列设计与侧链 packing 的分步流程;蛋白–小分子相互作用有专门的结构条件生成体系;片段连接、逆折叠、分子优化,各自都有成熟的技术路径。
每个方向都在进步,但它们之间几乎没有共享模型。其问题并不在能力,而在划分方式。
在现有体系中,小分子被当作“图问题”,多肽被当作“序列问题”,结构引导设计被当作“条件生成问题”。任务是按对象类型划分的,而不是按结构本质划分的。
从物理本质来看,蛋白质设计中的各类问题实际上遵循着相同的基本规律,包括原子间的相互作用、键长与键角的分布特征、局部几何结构的稳定性以及口袋空间的约束条件等,这些物理原理在小分子、多肽、蛋白片段乃至复合物体系中均保持一致;基于此,研究团队提出了一个核心问题:既然底层物理规律是统一的,为何模型却必须被割裂地开发和使用?
为了回答“能否用同一个模型处理不同分子生成任务?”这一问题,研究团队没有为小分子、多肽、片段设计不同网络,而是先做了一件更基础的事:把所有对象都抽象为同一种原子级表示。
无论是小分子图结构,还是蛋白三维结构,最终都被转化为:原子类型、原子三维坐标、化学键关系;模型则始终处理同一类结构节点与相互作用关系。在这样的设定下,表面看统一的是“任务”,其实真正统一的是“表示”。

图2. 建模单位重组架构
在整体架构设计中,不同任务虽拥有各异的输入形式,但其核心的生成模块却是共享的。这意味着任务之间的差异主要源于输入"条件"的不同,而非模型结构本身的分化;具体而言,无论是给定蛋白口袋来生成匹配的小分子、提供分子片段以完成结构扩展,还是输入功能或约束标签来生成符合条件的候选分子,这些看似不同的任务在架构图中并非各自独立的路径,而是通过条件控制机制汇入同一个生成核心进行处理。
这也就说明:任务不再是模型层面的分裂,而是输入层面的调控。
在模型训练的过程中,多种类型的数据被混合训练,而参数是共享的。这种多任务联合学习,使模型在不同任务之间建立结构上的联系,而不是孤立优化。

图3. 模型训练流程
研究思路的核心在于并非为每个具体问题开发专属工具,而是构建一个统一的模型来同时应对多种不同类型的任务。相应的,在应用过程中,还有另一个问题需要解决:不同生成任务,如何在同一个模型中被表达?
研究团队把所有生成问题都抽象为同一组基本要素:原子类型、原子坐标和键类型。区别不在于“做什么任务”,而在于哪些信息是固定的,哪些需要生成。

图4. 任务示意图
比如,在3D分子生成或基于结构的药物设计中,分子的原子类型、原子坐标和化学键类型都需要生成;在docking任务或者多构象生成任务中,原子类型和化学键类型被固定,但是原子坐标需要生成;在片段连接任务中,两个 fragment 是固定的,中间的 linker 需要生成;也就是说,任务的差异不再体现在模型结构层面,而体现在输入掩码的不同配置上。
这一步非常关键。它把原本彼此独立的任务,转化为同一种“条件生成”问题。模型看到的始终是同一种分子表示,只是生成区域不同。但统一表达还不够。不同任务在物理层面的约束差异也很明显。Docking、de novo 生成、片段生长,它们的构象自由度和噪声模式完全不同。在分子噪声的可视化图中可以看到,不同任务对应的分子分布在表示空间中形成相对独立的区域。这说明作者并没有强行用完全相同的噪声分布处理所有任务,而是在统一框架下引入了任务特异的噪声调度机制。
统一的是模型结构与表示方式,保留的是物理合理性。正是这两步——统一任务表达与可控噪声机制——让这个框架不仅在概念上成立,在技术上也站得住。
为了避免模型出现“什么都能做一点,但什么都不够好”的情况,研究团队进行了计算测评及应用场景测评。
在结构引导小分子设计(SBDD)任务中,模型需要在蛋白口袋约束下生成匹配的小分子。这类任务通常对三维几何、化学合理性、以及对接评分都非常敏感。结果显示,在分子图质量、物化性质、三维结构合理性等多项指标上,PocketXMol在大多数指标上处于领先位置。更关键的是,在“同时获得更优对接评分且三维结构有效”的分子比例上,它明显优于对比方法。这意味着模型生成的分子不仅“看起来好”,而且在结构匹配层面具有实质优势。
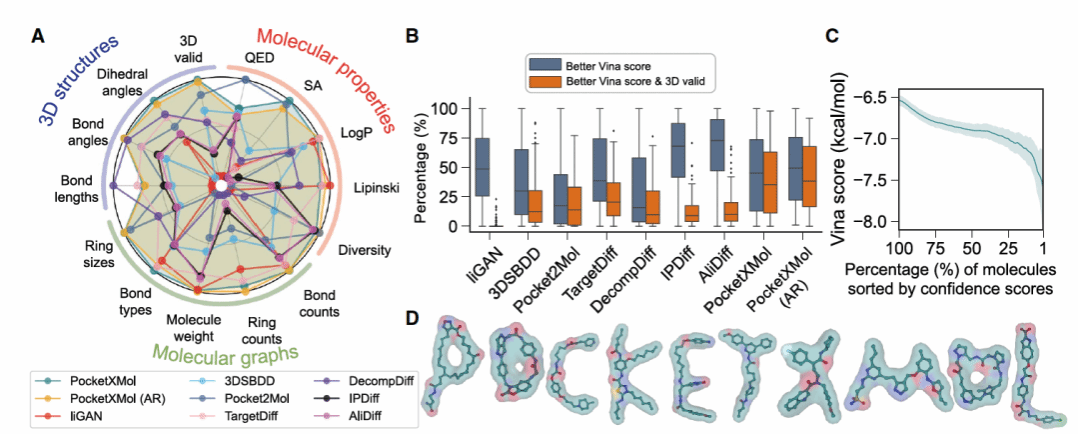
图5. 多任务测评结果1
在纯 3D 分子生成任务中,模型不依赖蛋白口袋约束,而是直接生成三维分子结构。任务结果显示,生成分子的几何统计分布与真实药物分子高度一致。这一点很重要,因为它说明模型学到的是分子结构的基本几何规律,而不是特定任务的技巧性偏置。
在 fragment linking 和 fragment growing 任务中,模型需要在“部分结构已知”的情况下补全剩余部分。这里的难点在于:既不能破坏固定片段的结构,又要保证连接部分在化学与几何上合理。结果显示,无论在已知片段姿态还是姿态扰动的情况下,PocketXMol都能保持较高的有效率与结构相似度。这与前面讲到的“任务特异噪声机制”是呼应的——统一框架并没有抹平不同任务的物理约束,反而通过统一表示让模型学到更稳健的结构规律。

图6. 多任务测评结果2
在分子优化任务中,模型被要求在保持结构相似性的前提下调整特定性质(例如 LogP)。实验结果显示,模型能够将分子性质稳定推向目标区间,同时避免剧烈结构偏移。这种“渐进式优化”能力,说明模型不仅能从零生成,也能在既有结构上做精细调整。
除了计算评测,PocketXMol也在实际场景下进行了测试。
在 caspase-9 抑制剂设计这一应用场景中,团队将模型生成的候选分子进行了合成与实验验证。部分分子在细胞水平和酶学实验中显示出显著抑制效果,并且结合位点预测通过突变实验得到支持。这意味着模型不仅在指标上表现优秀,而且能够产出具有真实生物活性的候选分子。
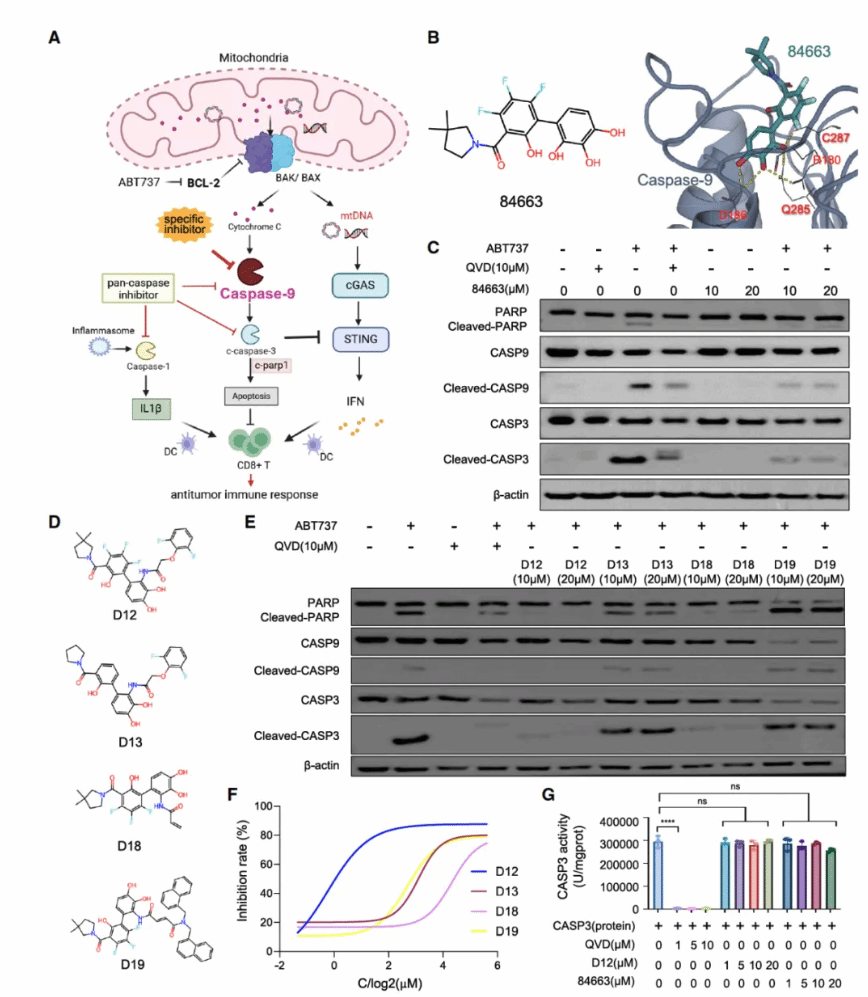
图7. 应用场景效果验证
测评结果表明PocketXMol并不是在某一个任务上“极端领先”,而是在多种不同约束、不同尺度、不同模态的任务下都保持稳定、高质量的表现。
统一,并没有带来能力塌缩。相反,多任务联合训练强化了模型对分子结构基本规律的理解,使它在不同场景下都能迁移应用。
如果说小分子部分证明了模型在传统药物设计流程中的能力,那么多肽部分考验的是一件更难的事情:同一个原子级生成框架,能否跨越分子类型?
为了回答这个问题,研究团队为PocketXMol设计了多个在传统建模体系中分属不同方法路径的任务,包括线性肽、环肽、含非天然氨基酸(NAAs)的设计,以及逆折叠任务,来测试模型的多肽设计能力。
1. 与传统多肽设计流程的区别
常规的多肽设计流程通常需要分步完成:首先生成主链骨架,再设计氨基酸序列,最后进行侧链构象优化;而PocketXMol采用了一种不同的策略,模型直接在原子层面同时生成主链与侧链的原子及键结构,待生成完成后,再根据已确定的侧链构象反向推断并标注对应的残基类型。
也就是说,模型并不是先预测氨基酸类别,再决定结构,而是先生成原子结构,再由结构推断氨基酸类型。
这一步本质上把“序列设计”问题转化成了“原子补全”问题。

图8. 多肽设计效果
结果显示,在蛋白–多肽复合物测试集上,生成的多肽在 Rosetta 结合能评分上与现有方法相当甚至更优。同时,氨基酸嵌入空间的聚类结构与真实氨基酸之间的替代关系高度一致,嵌入距离与 BLOSUM62 替代分数显著相关。
这说明模型不仅学到了几何结构,还学到了氨基酸之间的化学语义关系。
2. 与传统多肽设计方法对比
将PocketXMol与"RFdiffusion + ProteinMPNN + Rosetta"的经典流程进行对比时,研究团队发现前者在序列恢复率、结构RMSD以及Rosetta结合能三项指标上均表现更优;同时,其生成的多肽在二级结构比例分布上更接近真实测试集,表明模型未引入明显的结构偏置。换句话说,PocketXMol生成的多肽不仅结构合理,还在统计特征上与天然多肽高度相似。
3. 非天然氨基酸(NAAs)
由于模型直接在原子层面生成侧链,而不是先预测氨基酸类别,因此也能支持非天然氨基酸设计。
论文统计到 454 种不同的 NAA 侧链结构,覆盖不同尺寸与理化性质。并且在 Rosetta 结合能上,与标准氨基酸相比并未出现系统性劣势。
这一点是传统基于序列分类模型难以实现的。
4. PD-L1 结合肽实验验证
除了计算测评,模型在应用场景下也有突出表现。
团队设计了针对 PD-L1 的 10-residue 肽,并进行 SPRi 实验验证。结果显示:
• 15 条肽达到 ~10^-8 M 的结合亲和力
• 在去除训练集中 PD-L1 同源结构后,模型仍然能生成高亲和力肽
这一步证明了模型的泛化能力并非源于对相似结构的记忆。

图9. PD- L1结合肽实验验证
小分子与多肽两部分结果表明:原子级的统一表示,让"分子类型"本身不再构成建模的边界——模型所面对的不是割裂的"小分子任务"或"多肽任务",而是同一类原子与键的生成问题,区别仅在于输入约束条件的不同。
这一工作的真正突破体现在“表示层面”。传统方法中,小分子设计与多肽设计分属两套体系:小分子通常采用图模型,多肽则依赖序列模型或蛋白结构模型,不同任务之间需要单独设计处理流程。而本研究将问题抽象至原子级别,使所有任务统一转化为同一形式——在给定部分原子与键的条件下补全剩余结构。模型不再依据"分子类型"进行区分,仅识别"哪些字段是固定的"这一条件差异。
这一转变带来两个直接结果:一是多任务之间可共享结构规律,二是跨模态迁移成为自然产物而非额外设计。小分子与多肽的统一并非简单地"支持两种数据",而是通过统一表示从根本上消除了模态边界。从工程角度而言,这意味着无需为每种设计问题单独训练模型;从方法论角度而言,这意味着分子生成问题可能不再需要按类型进行划分。
PocketXMol的出现重新定义了分子生成问题:它不再被拆成“小分子设计”“多肽设计”“逆折叠”“片段连接”这些彼此分离的任务。在原子级表示下,它们只是不同的约束组合。
当任务差异从“模型结构差异”变成“输入掩码差异”时,多模态不再是拼接,而是自然统一。
当然,这并不意味着从此所有分子设计问题都能一网打尽。数据规模、训练分布、物理约束,这些问题依然存在。但至少在表示层面,研究团队提供了一种更简洁的答案。
如果说前几年的关注点在于模型能力在单一方向上的突破,那么这篇文章展示的,是生成问题在结构层面的整合。它不执着于某个任务的最优解,而是一次框架的重构。而框架的改变,往往比单点性能提升更值得关注。


